The new EU regulation on health technology assessment: Major change or minor adjustment?
By HTA Quarterly
.jpg?h=3299&iar=0&w=5000&hash=03BAD8086A4A080FF9D341B726210C94)
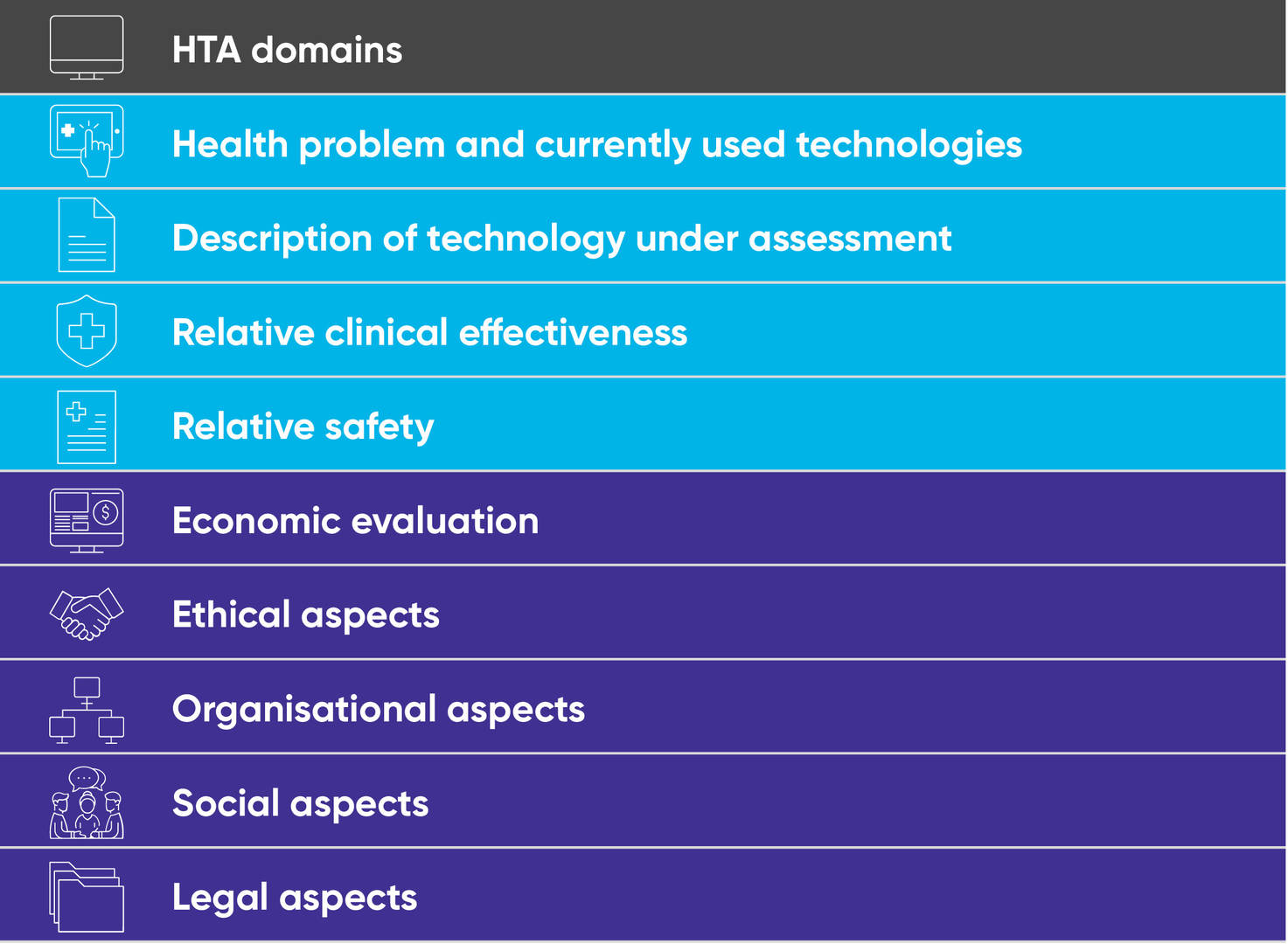
After many years of fruitful voluntary collaboration of EU member states in HTA, through the EUnetHTA Joint Action and its predecessors, the EU finally passed a regulation in December 2021 to pave the way for more formal collaboration in HTA across the member states. (See related article here.) The new proposals, which came into force on January 11, 2022, focus on the “clinical domains” of HTA, including the assessment of relative efficacy or effectiveness (shown in light blue below). All the other relevant “non-clinical” domains including economic evaluation, which also need to be considered for a holistic assessment of potential gains from adopting new technologies in the local setting, will remain the responsibility of individual member states.
Specifically, under the new regulation, member states’ HTA bodies are expected to conduct joint clinical assessments (JCAs) of new medicines and some high-risk medical devices. In addition, member states will engage in joint scientific consultations (JSCs) to advise health technology developers on clinical study designs for generating appropriate clinical evidence. The whole process will be supported by more “horizon scanning” at the EU level, to alert member states to important new health technologies in the pipeline.
The main motivations for the change are twofold. First, there may currently be some wasted time and effort, due to the HTA bodies of several member states conducting the same systematic review of the clinical evidence. Under the new regulation, the JCA, once produced, will be available for use by all member states. Secondly, the change might encourage more member states to use HTA in their adoption decisions for new technologies, if in the past the need to produce a local clinical assessment was a major barrier to including HTA in their decision-making processes.
As there are already many published articles on policy implications, as well as advantages and disadvantages of the new regulation, we focus here on its likely impact on health technology developers, especially with regard to their responsibilities for the provision of data or evidence submissions to national HTA bodies. In particular, we present some thoughts on the opportunities health technology developers may have to influence the focus of HTA in relation to their own products, or at an industry level through their trade associations. Like many (all) changes in the EU, this change will not happen overnight. The Member State Coordination Group on HTA (HTACG), consisting of national representatives, has been assembled to hammer out the details, prior to full implementation of the regulations by January 2025, with the assessment of oncology products as a first area of focus, since this was identified by policy makers as having the highest unmet need for a unified assessment.
One consequence we are already aware of is that a technology developer will only have to submit the data for the JCA once and can resist requests from individual member states for the same data. This may seem like an important change, potentially reducing companies’ workloads, but a concession was made during the agreement of the regulation that the existence of a JCA report should not affect the discretion of member states to carry out their own assessments of the technologies concerned. Member states will be able to perform complementary clinical analyses of health technologies for which a JCA report is available, if these are necessary for their national HTA process. Member states will also be able to perform complementary clinical analyses relating, inter alia, to patient groups, comparators, or outcomes other than those included in the JCA report.
Member states can also apply a different methodology, if that methodology would be required in the overall national HTA process of the member state concerned. A recent paper by Kisser et al identified four important areas where there are differences between the current EUnetHTA JCA methodology and the current German approach to HTA: 1) applying a view of differing treatment algorithms and definition of corresponding subpopulations/respective comparators; 2) clinical relevance of surrogate/intermediate endpoints; 3) tolerance to accept clinical study interventions not used for marketing authorisation; and 4) different operationalisation and/or weighting of individual safety endpoints leading to different safety conclusions. Therefore, member states will still be able and likely to ask the health technology developer to submit additional necessary data or analyses to meet diverging needs.
Thus, our assessment is that the new regulation is likely to have very little impact on companies’ responsibilities, including the need to provide the appropriate evidence to individual member states, despite providing clinical data for the JCA. Having said that, much, however, depends on the likely scope of the JCAs. They may be restricted to a systematic review of head-to-head randomised controlled trials, using the clinical endpoints reported in the clinical studies reviewed. If so, they will focus on an important, but limited, component of the total evidence required to make recommendations on the use of health technologies. On the other hand, the JCAs might be quite broad, involving extrapolation beyond the observation period in the clinical trials, commenting on the validity of certain surrogate endpoints for predicting long-term outcomes, and using evidence beyond that found in head-to-head clinical trials (such as real-world evidence and observational studies) to assess how the new technology compares with relevant alternatives.
In another paper, Drummond et al point out that expanding the role of JCAs raises numerous methodological issues, on which there is not a clear consensus among member states. The critical issues include the choice of survival models, the validation procedures for surrogates, and the ways of addressing the biases when attempts are made to make comparisons outside of those seen in head-to-head randomised clinical trials. The issue of making indirect comparisons of the relative effectiveness of health technologies has been discussed in a recent draft methodological guideline produced by EUnetHTA. This acknowledges that there are methods to make comparisons of the relative effectiveness of technologies when head-to-head randomised controlled trials (RCTs) do not exist, but that these are highly dependent on the quality of the evidence network available. (For example, does the evidence network include connected RCTs or mainly non-randomised studies? Are individual patient data available to help in addressing confounders, or are only aggregate estimates of effectiveness available from the clinical studies?) The guideline concludes that “In many cases the conditions will not be ideal for the use of any of the methods presented in this guideline to produce unbiased assessments of relative effectiveness.”
A related issue concerns the fact that the comparators to a new technology, and the importance of various clinical outcomes, may vary from one member state to another. Another recent EUnetHTA draft guideline document on the scoping of HTAs points out that the scope of HTAs reflects the policy questions in the different healthcare systems in which the HTA is used. These are reflected in the PICO framework (Population, Intervention, Comparator, Outcome), which may also significantly differ from one member state to another. Therefore, the guideline envisages that as part of the development of the JCA, a survey of member states would be conducted to identify the relevant PICOs. However, it recognises that the number of PICOs explored in the JCA should be minimised as much as possible, thereby implying that some member states’ needs may not be fully met by the JCA.
These issues need to be explored by the HTACG in the context of individual JCAs. However, given these challenges, it would be no surprise if JCAs are often limited in scope, representing a small part of the whole HTA evidence package. Therefore, technology developers should anticipate the need to provide additional data and analyses to individual member states to order to meet their specific needs.
So, what can technology developers do to influence the implementation of the new regulation? Our message is that, in the context of developing an evidence package for a particular product, the company’s destiny remains largely in its own hands. It is unlikely that in many, if any, cases the JCA will be so conclusive and so damning that the marketing opportunities for a product will be seriously curtailed. That is not to say that technology developers should not take the JCAs seriously. Obviously, all the relevant data should be made available (RCTs or other studies, if required) and progress in the JCA monitored to guard against the emergence of any misleading arguments or findings. However, we believe that the market access needs and opportunities for new products will largely remain as at present, in particular in relation to how technology developers relate to the HTA bodies in the member states.
Finally, the team performing the JCA can ask the technology developer for more data if it feels that the data provided do not enable it to make an adequate assessment. The likelihood of this happening also depends on the scope of the JCA, but it is difficult to imagine what additional data could be provided quickly. For example, if the request is to, say, provide data on overall survival from trials of anti-cancer drugs, as opposed to data on progression-free survival, this could take months or years to generate. We feel that it is unlikely that all member states would be willing to wait that long for the assessment, and, in any case, may see this as a member state activity, better handled through country-level managed entry agreements. Of course, over time the data needs for the JCAs will be determined by the JSCs, so problems relating to the inadequacies of the clinical evidence are likely to diminish over time, although for the first few years after the implementation of the new regulations in January 2025, the products that are the subject of JCAs will not have had the benefit of going through the JSC process.
Given that the discussions within the HTACG are likely to influence the implementation of both JCAs and JSCs, it is important that technology developers monitor progress of the discussions, perhaps through industry associations, since an important part of the HTACG’s remit is to engage with stakeholders. There are a number of considerations here and it is not our role to determine a unified industry strategy. However, one important dynamic will be the participation of the medical device industry in these discussions. Medical device companies vary greatly in size and in their ability to produce evidence on the effectiveness and cost-effectiveness of their products. Therefore, medical device companies are currently more concerned about the new EU regulation than are pharmaceutical companies. We believe that it is better for technology developers to be constructively critical, rather than to dismiss arguments out of hand. We feel that society has reached the point where it expects there to be a discussion of the clinical and cost-effectiveness of new health technologies before they are made available. Therefore, the new EU regulation is appropriate, and all parties should work together to ensure that it is effective.
However, one aspect that industry stakeholders might guard against is the adoption of absolutist positions. For example, there are some who believe that head-to-head RCTs are the only form of reliable evidence on relative effectiveness of health technologies, that the use of all surrogate endpoints is dubious, or that real-world data are so fraught with bias that they are unusable. While we are aware that such views exist, it would be unfortunate if, through the discussions of the HTACG, they gain some level of EU-wide acceptance. In our view a more nuanced position should prevail. (See Table 1 below.)
Table 1. Differences between absolutist and nuanced positions on key methodological issues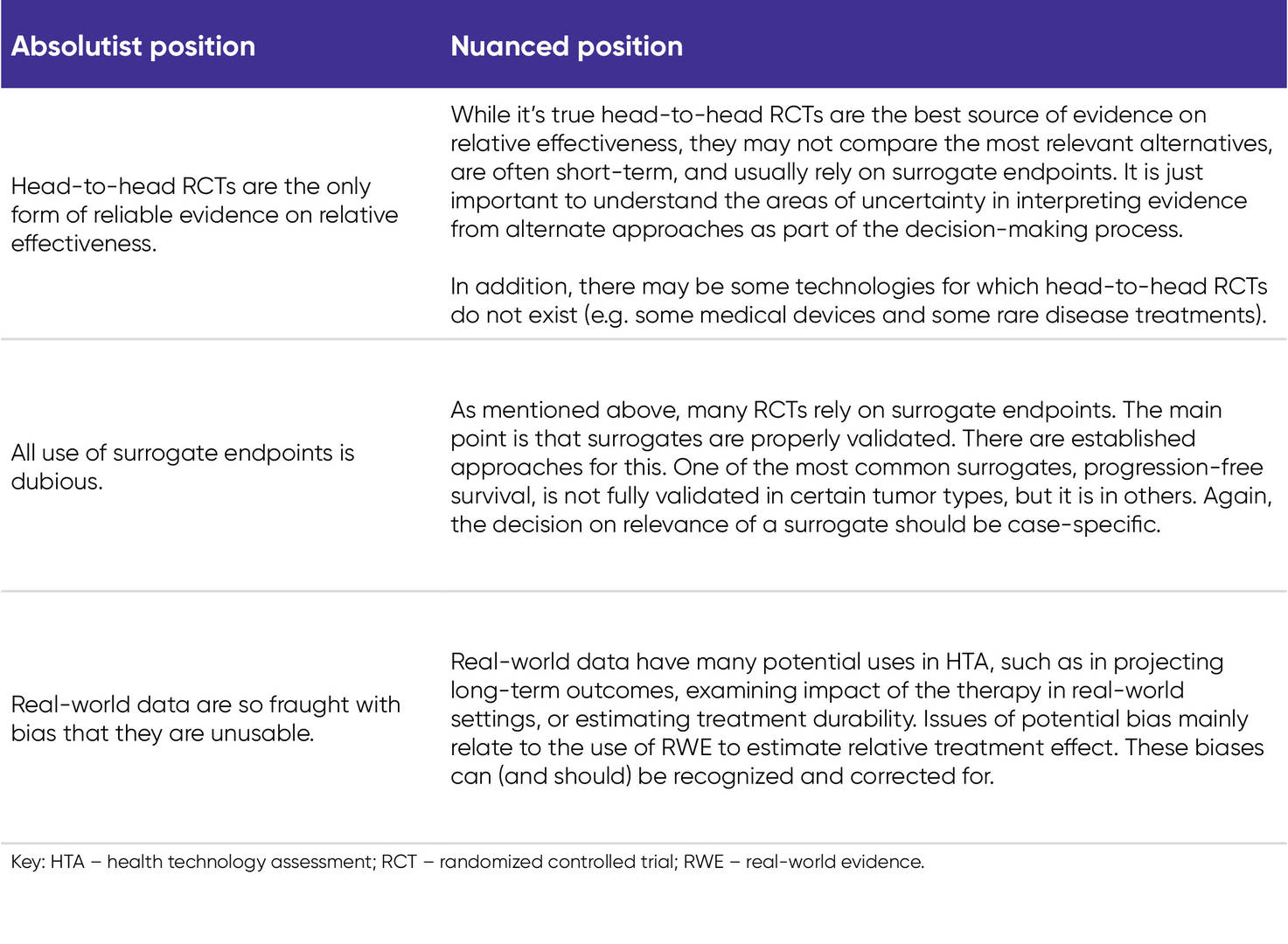
In conclusion, although the new EU HTA regulation represents an important change, it is unlikely to pose an important barrier on the market access of new technologies. However, it is important that technology developers are at least aware of the likely impact of the changes, so that they can adjust to them appropriately.
For further information on this topic, please contact us, or explore our global market access solutions.
Sources
- Drummond M, Tarricone R, Torbica A. European Union regulation of health technology assessment; what is required for it to succeed? Eur J Health Econ. 2022; 23(6):913-915.
- Kisser A, Knieriemen J, Fasan A, et al. Towards compatibility of EUnetHTA JCA methodology and German HTA: a systematic comparison and recommendations from an industry perspective. Eur J Health Econ. 2022;23:863-878.
- EUnetHTA 21. Practical guideline scoping process. Version 0.3. February 5, 2022. Accessed July 28, 2022. https://www.eunethta.eu/wp-content/uploads/2022/05/4.2-Scoping-Process-Practical-Guideline-version-for-public-consultation-v0.3.pdf?x69613
- EUnetHTA 21. D4.3.2: Direct and indirect comparisons. Version 0.3. May 2, 2022. Accessed July 28, 2022. https://www.eunethta.eu/wp-content/uploads/2022/05/D4.3.2_GL_Comparisons_for-public-consultation.pdf?x69613=

About the author
Michael Drummond, PhD
Professor Emeritus and former Director of the Centre for Health Economics at the University of York, UK
Michael Drummond, PhD, is Professor Emeritus and former Director of the Centre for Health Economics at the University of York in the United Kingdom. His main field of interest is in the economic evaluation of healthcare treatments and programmes.
Dr. Drummond is the author of 2 textbooks on healthcare economics and more than 700 scientific papers. He served as President of the International Society of Technology Assessment in Health Care and the International Society for Pharmacoeconomics and Outcomes Research. He was a member of the Steering Group for the 2020–2022 NICE Methods Review. He has advised several governments on the assessment of health technologies and chaired one of the Guideline Review Panels for the National Institute for Health and Care Excellence (NICE) in the UK.
In October 2010, Dr. Drummond became a member of the National Academy of Medicine in the United States. He is currently co-editor-in-chief of Value in Health and has been awarded 3 honorary doctorates from City University (London), Erasmus University (Rotterdam), and the University of Lisbon.

About the author
Thomas Mittendorf, PhD
Vice President, Real-World Evidence & HTA Support
Xcenda / AB Consulting
Thomas Mittendorf, PhD, is Vice President and Practice Area Lead of Real-World Evidence and Health Technology Assessment (HTA) Support for AB Consulting. In this role, Dr. Mittendorf heads a team of 100+ that conducts International HTA Market Access, German HTA Support, and Real-World Evidence in the US as well as Real-World Evidence in Europe.
Dr. Mittendorf and his team are dedicated to facilitating a deeper understanding of health policy and regulatory issues across a variety of areas and technologies, including oncology, neurology, cell and gene therapies, and orphan indications. A former editor-in-chief of ISPOR Connections, he has authored articles appearing in more than 120 scientific publications. Today he sits on several editorial boards and acts as a regular contributor and reviewer for multiple peer-reviewed journals.
Dr. Mittendorf earned his PhD and Master of Science degrees from Leibniz University of Hannover, Germany. He also has held an adjunct professor role there in health economics and health system research at the Center for Health Economics.